

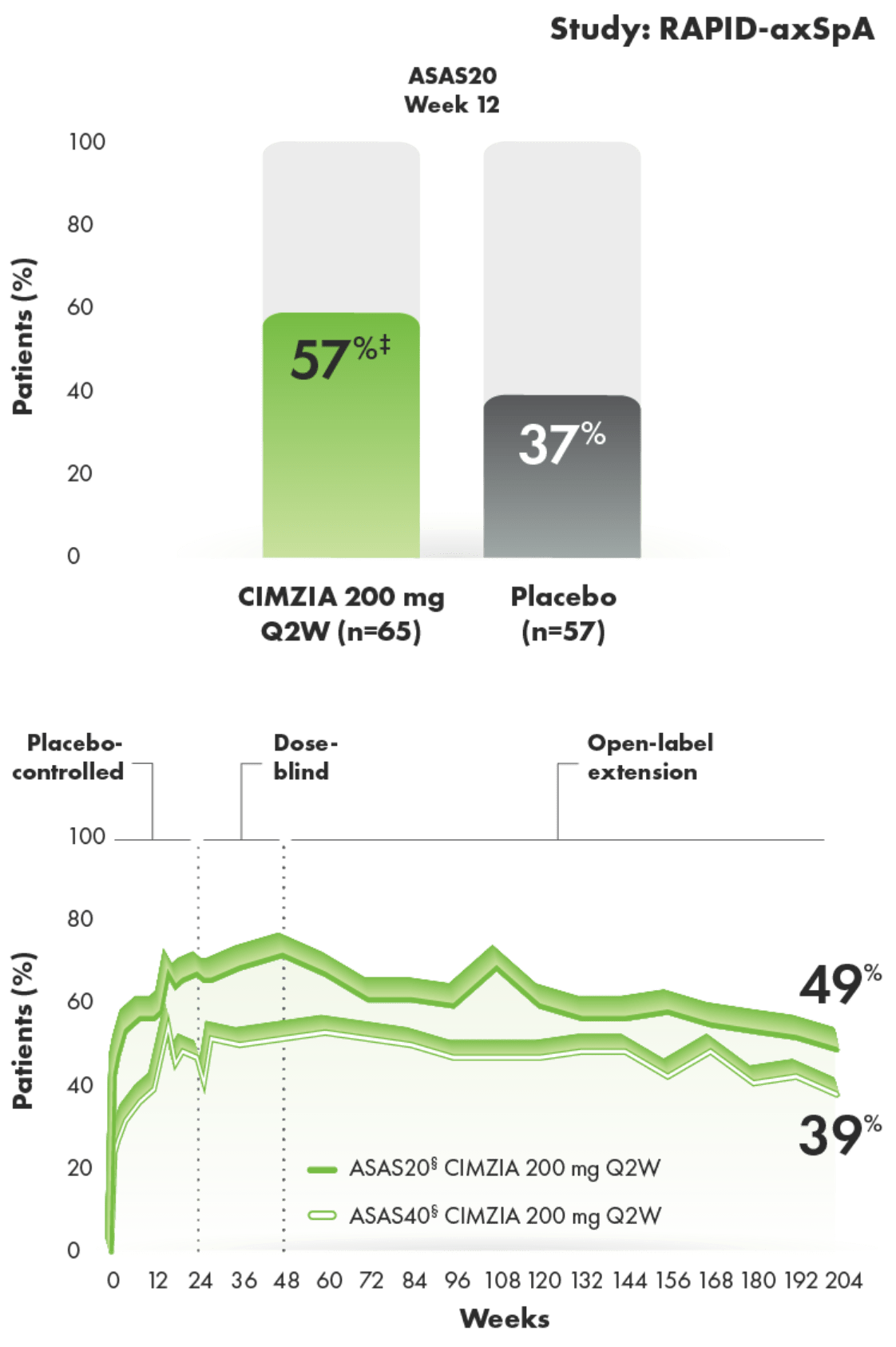
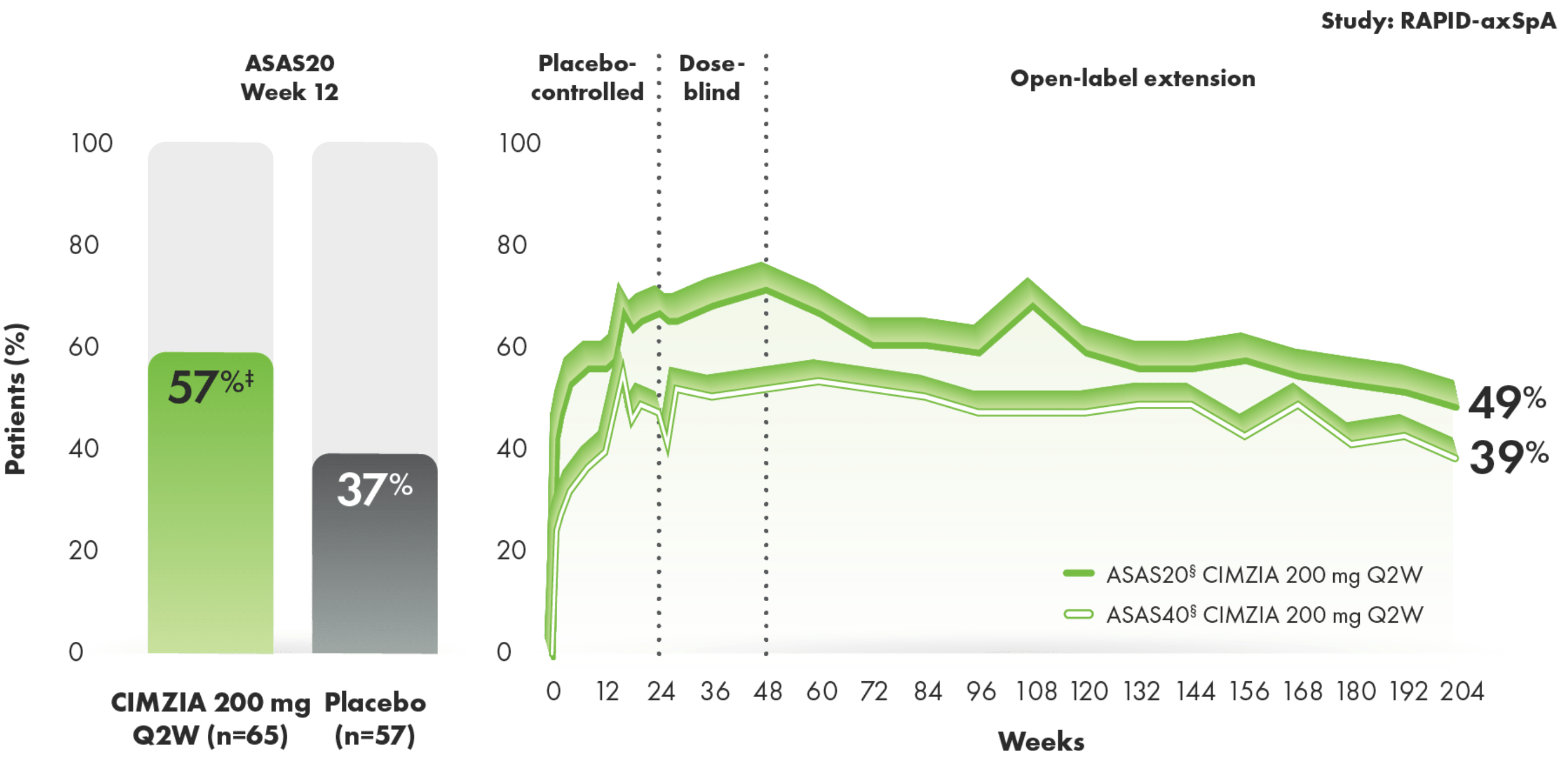
Sustained improvement in signs and symptoms of AS over 4 years
ASAS20/40 response rates in an AS subpopulation through Year 41-3*†
SOME EXPERIENCED ASAS20 RESPONSES AS EARLY AS 1 TO 2 WEEKS AFTER STARTING CIMZIA1,2
- Placebo rates at Week 242
- ASAS20: 33%
- ASAS40: 16%
- ASAS criteria measure improvements in pain, function, morning stiffness, and patients’ overall assessment of disease activity
- Limitations of OLE data include potential bias due to open-label treatment and lack of long-term placebo control beyond Week 24‖
*The same patients may not have responded at each time point.
†RS-NRI: randomized set nonresponder imputation.
‡ASAS20 at Week 12 in the AS subpopulation was a prespecified analysis of the primary efficacy end point. Nominal P value.
§ASAS20 and ASAS40 at Week 204 were prespecified secondary end points.
||Line graph to Week 204 represents patients who were randomized initially to CIMZIA 200 mg Q2W.
RAPID-axSpA2,4
The RAPID-axSpA trial (nr-axSpA and AS patients) was a 204-week, Phase 3, multicenter, randomized, double-blind, placebo-controlled study of 325 patients aged ≥18 years who had either nr-axSpA (n=147) or AS (n=178). Patients were randomized to CIMZIA 200 mg Q2W (n=111), CIMZIA 400 mg Q4W (n=107), or placebo (n=107). CIMZIA-treated patients were given a loading dose of 400 mg of CIMZIA at Weeks 0, 2, and 4. The 24-week, double-blind, placebo-controlled period was followed by a dose-blind phase. CIMZIA-treated patients continued to receive their original dose regimen after Week 24 (200 mg Q2W [n=105] or 400 mg Q4W [n=98]). Placebo-treated patients who did not achieve an ASAS20 response at Weeks 14 and 16 were re-randomized to CIMZIA 200 mg Q2W (n=27) or CIMZIA 400 mg Q4W (n=29), and those who completed therapy to Week 24 were re-randomized to CIMZIA 200 mg Q2W (n=20) or CIMZIA 400 mg Q4W (n=21). An open-label period followed at Week 48 and continued to Week 204. Note: At the conclusion of the RAPID-axSpA study, CIMZIA was not yet approved for treating patients with nr-axSpA.
AS, ankylosing spondylitis; ASAS, Assessment of SpondyloArthritis international Society; nr-axSpA, non-radiographic axial spondyloarthritis; OLE, open-label extension; Q2W, every 2 weeks; Q4W, every 4 weeks.
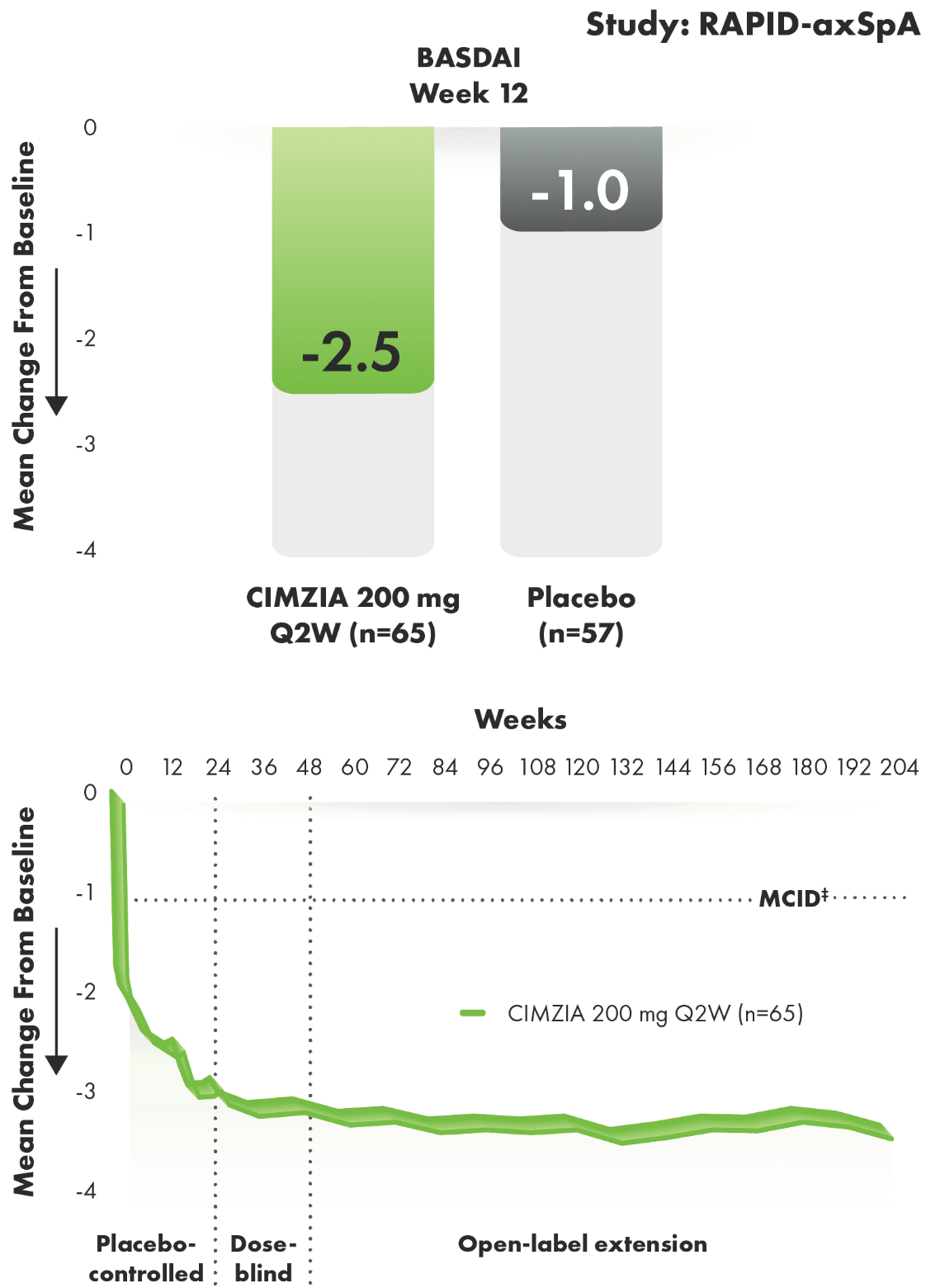
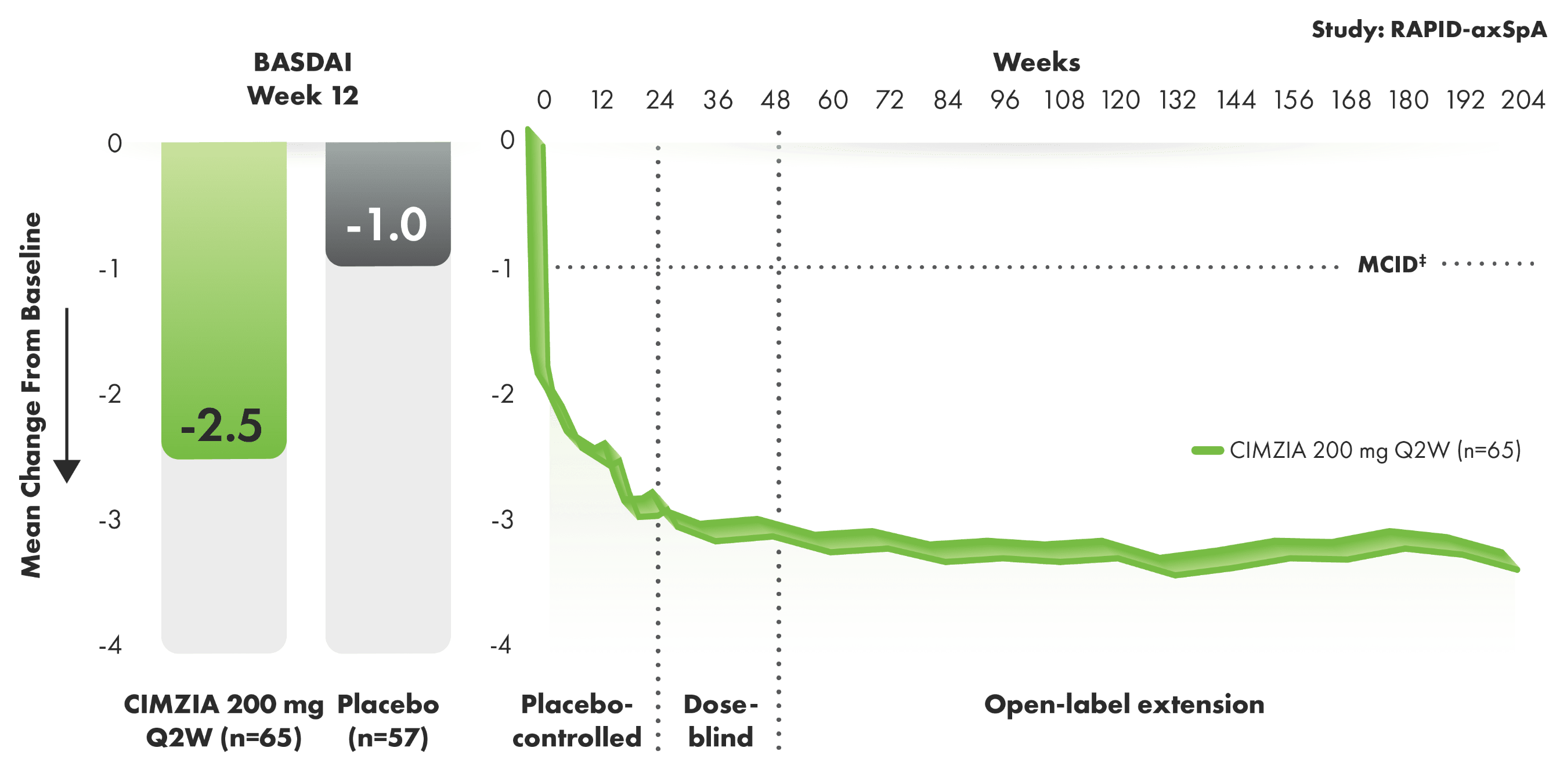
CIMZIA patients experienced consistent improvement in disease activity through
4 years1-3 CIMZIA patients experienced consistent improvement in disease activity through 4 years1-3
BASDAI improvement from baseline through Year 4 in AS1-3*†
BASDAI components of disease activity


- Limitations of OLE data include potential bias due to open-label treatment, lack of long-term placebo control beyond Week 24, and potential enrichment of population with responders
- The BASDAI consists of a 0 to 10 NRS, which is used to answer questions pertaining to major symptoms of AS5
- Baseline BASDAI score was 6.52 for CIMZIA patients and 6.44 for placebo patients2,3
*RS-LOCF: randomized set last observation carried forward.
†Change from baseline in BASDAI scores in the AS subpopulation was a prespecified end point. Nominal P value.
‡MCID was defined as a reduction of 1 unit on the NRS.3
RAPID-axSpA2,4
The RAPID-axSpA trial (nr-axSpA and AS patients) was a 204-week, Phase 3, multicenter, randomized, double-blind, placebo-controlled study of 325 patients aged ≥18 years who had either nr-axSpA (n=147) or AS (n=178). Patients were randomized to CIMZIA 200 mg Q2W (n=111), CIMZIA 400 mg Q4W (n=107), or placebo (n=107). CIMZIA-treated patients were given a loading dose of 400 mg of CIMZIA at Weeks 0, 2, and 4. The 24-week, double-blind, placebo-controlled period was followed by a dose-blind phase. CIMZIA-treated patients continued to receive their original dose regimen after Week 24 (200 mg Q2W [n=105] or 400 mg Q4W [n=98]). Placebo-treated patients who did not achieve an ASAS20 response at Weeks 14 and 16 were re-randomized to CIMZIA 200 mg Q2W (n=27) or CIMZIA 400 mg Q4W (n=29), and those who completed therapy to Week 24 were re-randomized to CIMZIA 200 mg Q2W (n=20) or CIMZIA 400 mg Q4W (n=21). An open-label period followed at Week 48 and continued to Week 204. Note: At the conclusion of the RAPID-axSpA study, CIMZIA was not yet approved for treating patients with nr-axSpA.
CIMZIA provided AS patients with meaningful improvement in back pain
Total and nocturnal spine pain improvement from baseline through Week 243§||
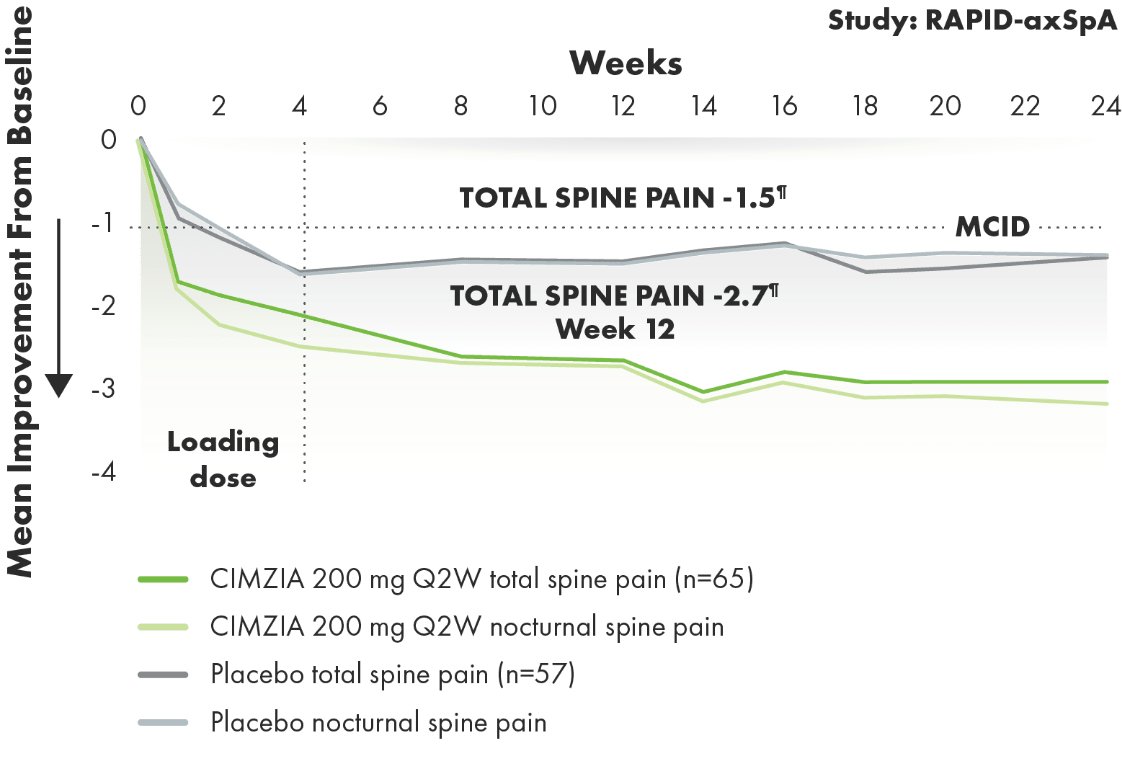
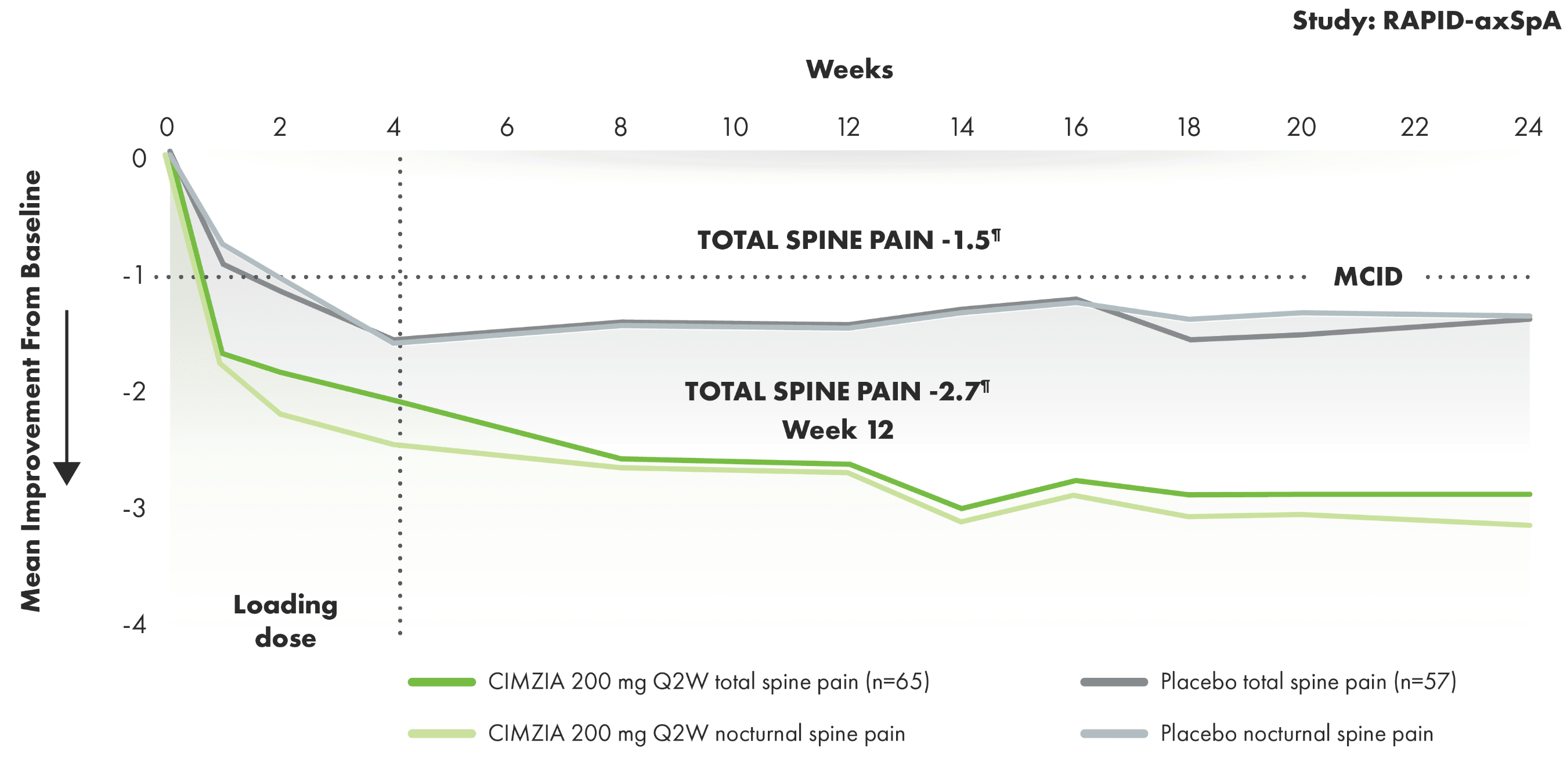
- Total spine pain is a component of ASAS response criteria measured by a 10-point NRS6
- Results were similar for both total spine pain and nocturnal back pain through Week 243
- MCID was defined as a reduction of 1 unit on the NRS3
- Baseline total and nocturnal spine pain scores were 7.0 and 6.7 for CIMZIA patients and 7.3 and 7.3 for placebo patients, respectively3
§FAS-LOCF: full analysis set last observation carried forward.
||Nocturnal back pain was evaluated by a 10-point NRS.
¶Change from baseline in total spine pain scores in the AS subpopulation was a prespecified end point. Nominal P value.
Significant clinical improvements in physical function1-3
BASFI improvement through Week 241-3#
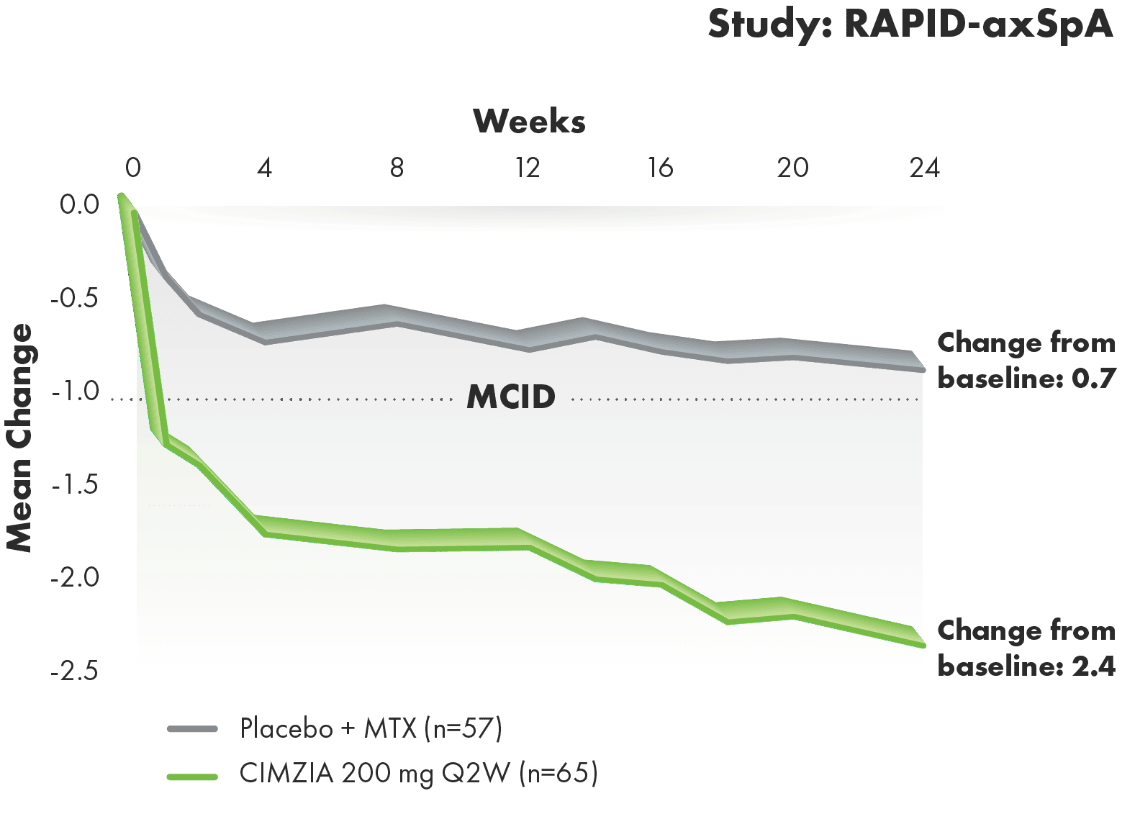
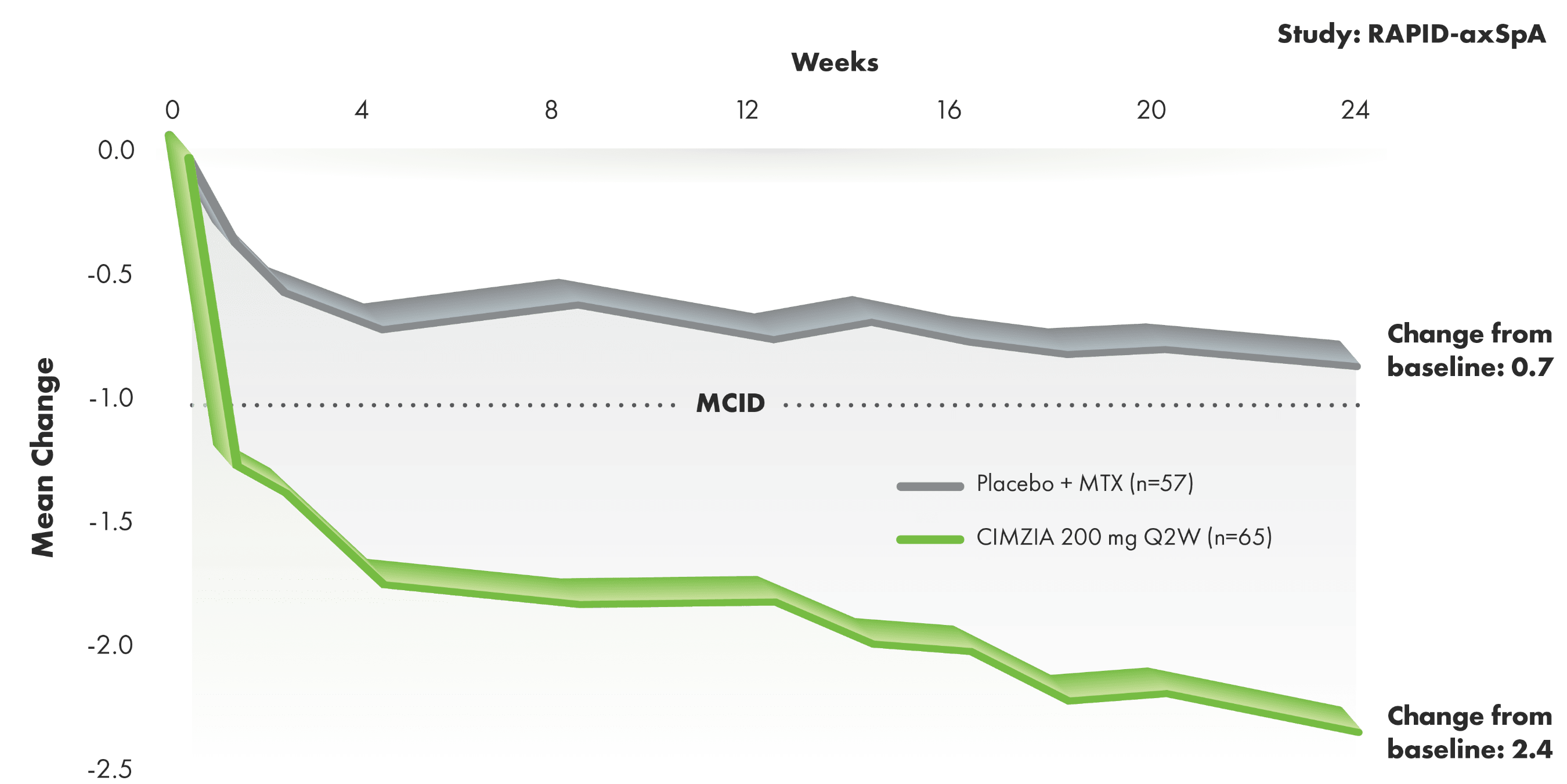
AT 24 WEEKS, CIMZIA OFFERED A 2.7-FOLD GREATER PERCENT IMPROVEMENT IN FUNCTIONING FROM BASELINE VS. PLACEBO2
- Change from baseline in BASFI scores in the AS subpopulation represents improvements in patients’ ability to perform certain activities of daily living, which was a prespecified secondary end point. Nominal P value
- MCID was defined as a reduction from baseline of -1 unit on the BASFI NRS in RAPID-axSpA
#RS-LOCF: randomized set last observation carried forward.
AS, ankylosing spondylitis; ASAS, Assessment of SpondyloArthritis international Society; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; BASFI, Bath Ankylosing Spondylitis Functional Index; MCID, minimum clinically important difference; MTX, methotrexate; nr-axSpA, non-radiographic axial spondyloarthritis; NRS, numeric rating scale; OLE, open-label extension; Q2W, every 2 weeks; Q4W, every 4 weeks.
Safety profile through 24 weeks in the AS subpopulation of RAPID-axSpA1,3
Incidence of TEAEs through Week 241,3
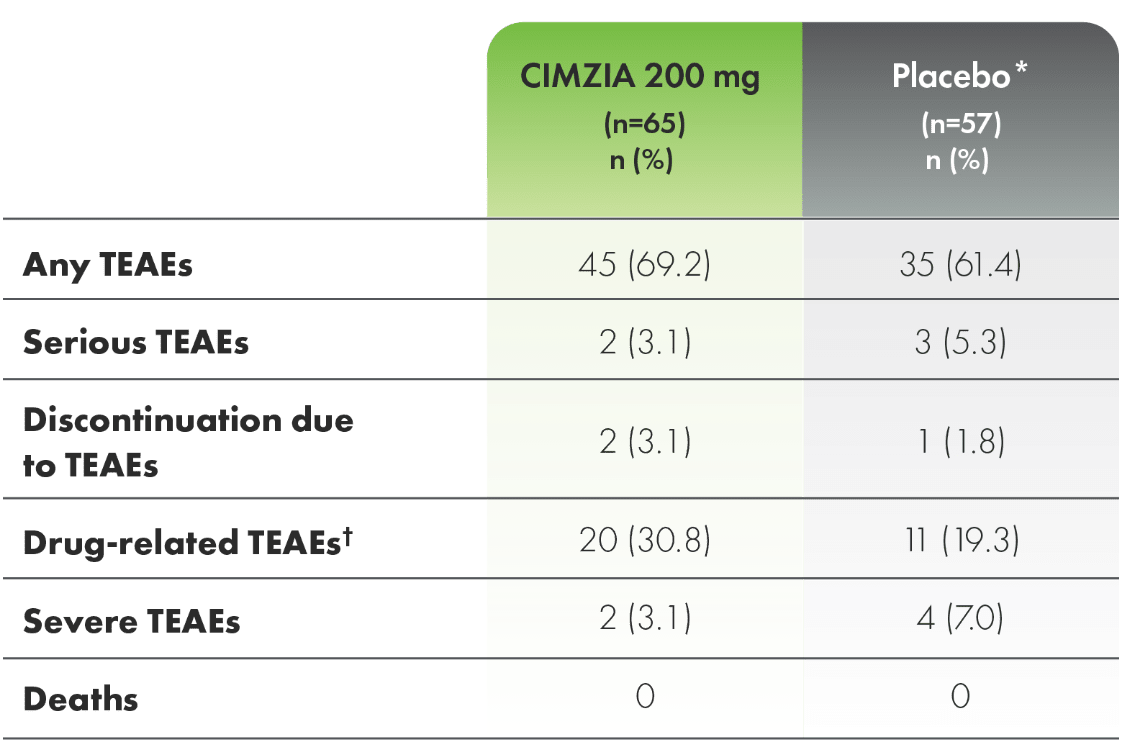
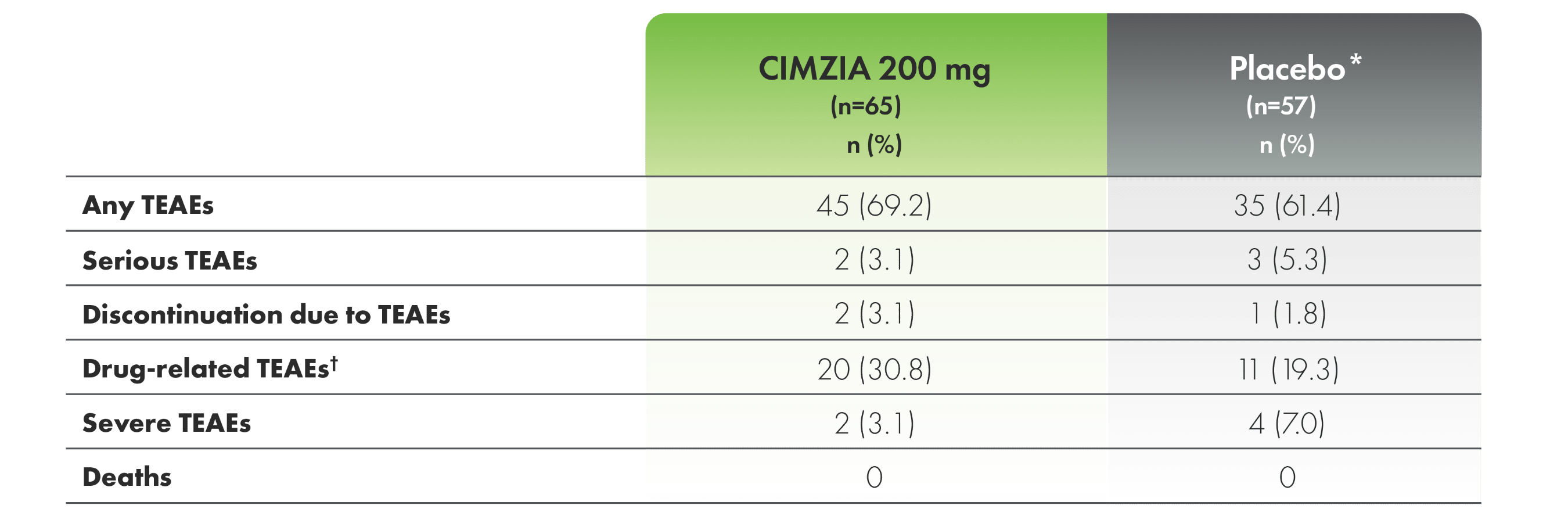
*For the entire placebo group, CIMZIA data from placebo subjects are not utilized.
†Drug-related TEAEs are those with a relationship of “related,” “possibly related,” or those with missing responders.
Adverse events in ≥5% of patients (CIMZIA 200 mg) through Week 241,3
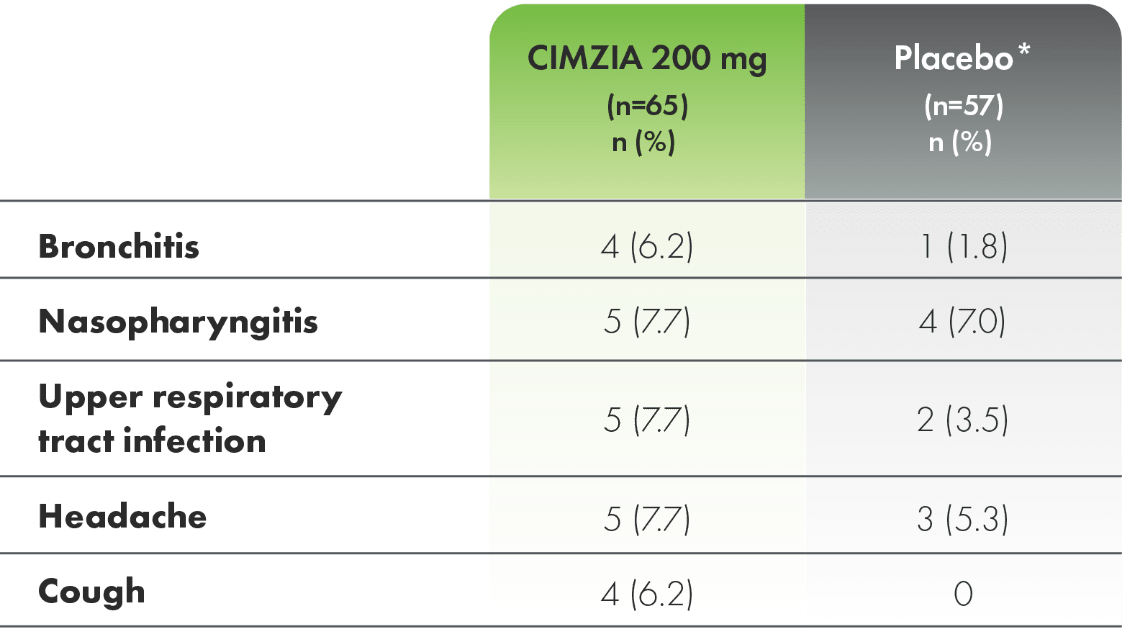
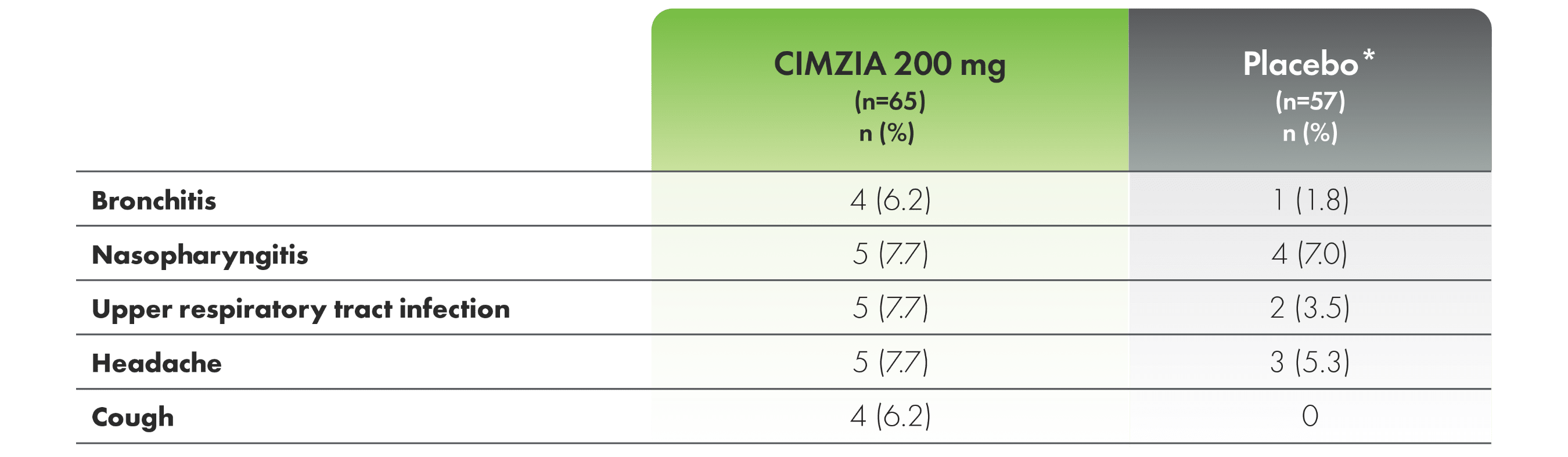
*For the entire placebo group, CIMZIA data from placebo subjects are not utilized.
AS, ankylosing spondylitis; TEAE, treatment-emergent adverse event.
Fc, fragment crystallizable; PEG, polyethylene glycol; TNF, tumor necrosis factor.